现代材料检测技术.docx
《现代材料检测技术.docx》由会员分享,可在线阅读,更多相关《现代材料检测技术.docx(15页珍藏版)》请在冰点文库上搜索。
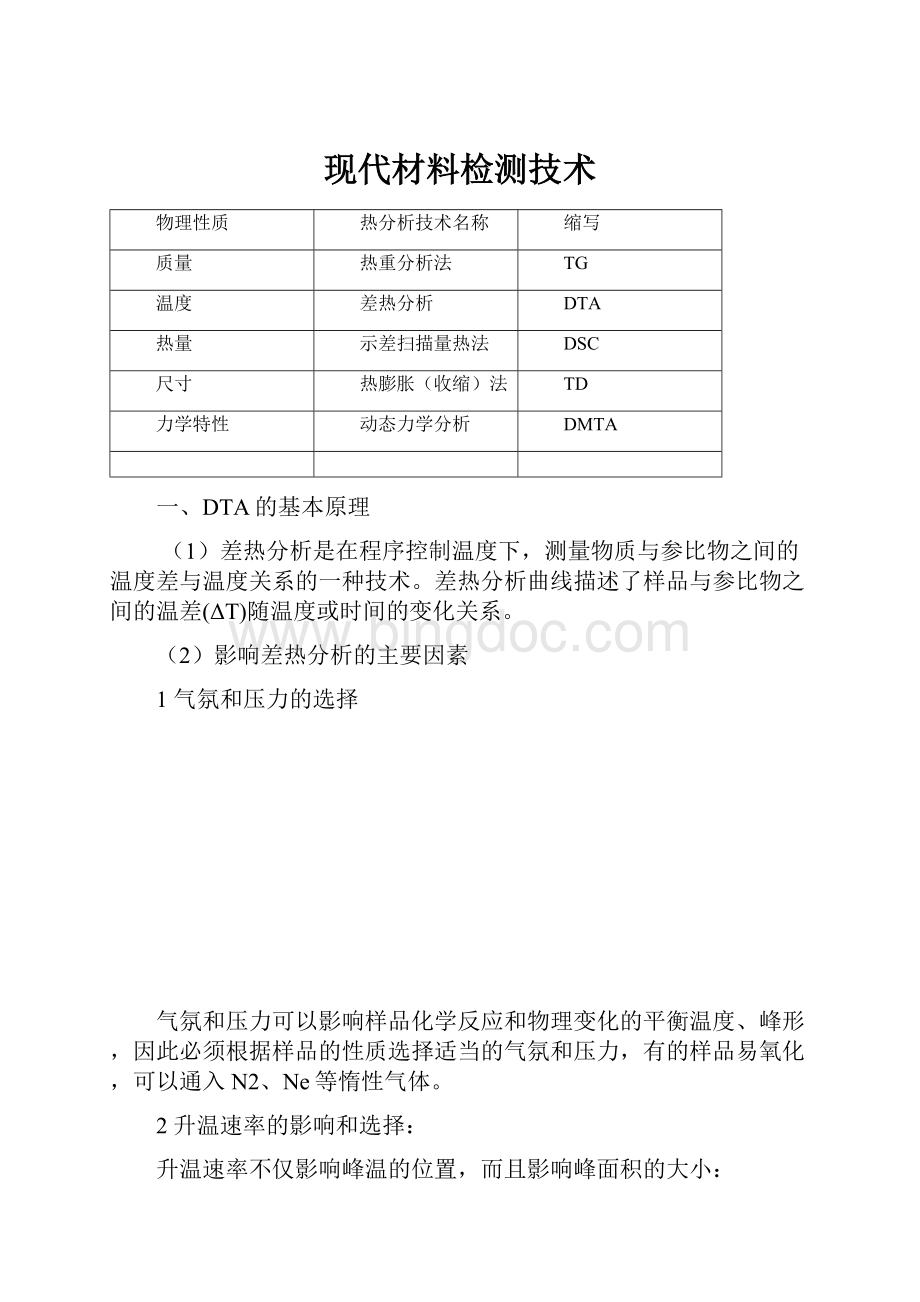
现代材料检测技术
物理性质
热分析技术名称
缩写
质量
热重分析法
TG
温度
差热分析
DTA
热量
示差扫描量热法
DSC
尺寸
热膨胀(收缩)法
TD
力学特性
动态力学分析
DMTA
一、DTA的基本原理
(1)差热分析是在程序控制温度下,测量物质与参比物之间的温度差与温度关系的一种技术。
差热分析曲线描述了样品与参比物之间的温差(ΔT)随温度或时间的变化关系。
(2)影响差热分析的主要因素
1气氛和压力的选择
气氛和压力可以影响样品化学反应和物理变化的平衡温度、峰形,因此必须根据样品的性质选择适当的气氛和压力,有的样品易氧化,可以通入N2、Ne等惰性气体。
2升温速率的影响和选择:
升温速率不仅影响峰温的位置,而且影响峰面积的大小:
快的升温速率下峰面积变大,峰变尖锐。
使试样分解偏离
平衡条件的程度也大,易使基线漂移,并导致相邻两个峰重
叠,分辨力下降。
慢的升温速率,基线漂移小,使体系接
近平衡条件,得到宽而浅的峰,也能使相邻两峰更好地分
离,因而分辨力高。
但测定时间长,需要仪器的灵敏度高。
升温速率对高岭土差热曲线的影响:
升温速率越大,峰形越尖,峰高也增加,峰顶温度也越高
3试样的预处理及粒度
试样用量大,易使相邻两峰重叠,降低了分辨力。
一般尽可能减少用量,最多大至毫克。
样品的颗粒度在100目~200目左右,颗粒小可以改善导热条件,但太细可能会破坏样品的结晶度。
对易分解产生气体的样品,颗粒应大一些。
参比物的颗粒、装填情况及紧密程度应与试样一致,以减少基线的漂移。
试样量越大,差热峰越宽,越圆滑。
其原因是因为加热过程中,从试样表面到中心存在温度梯度,试样越多,梯度越大,峰也就越宽。
4参比物的选择
要获得平稳的基线,要求参比物在加热或冷却过程中不发生任何变化,在整个升温过程中其比热、导热系数、粒度尽可能与试样一致或相近。
常用α-三氧化二铝Al2O3)或煅烧过的氧化镁(MgO)或石英砂作参比物。
如果试样与参比物的热性质相差很远,则可用稀释试样的方法解决;常用的稀释剂有SiC、铁粉、Fe2O3、玻璃珠Al2O3等。
5纸速的选择
在相同的实验条件下,同一试样如走纸速度快,峰的面积大,但峰的形状平坦,误差小;走纸速率小,峰面积小。
因此,要根据不同样品选择适当的走纸速度。
不同条件的选择都会影响差热曲线,除上述外还有许多因素,诸如样品管的材料、大小和形状、热电偶的材质以及热电偶插在试样和参比物中的位置等。
二.热重分析(ThermogravimetricAnalysis)
(1)热重法(Thermogravimetry,TG)是在程序控温下,测量物质的质量与温度或时间的关系的方法,通常是测量试样的质量变化与温度的关系。
热重分析的结果用热重曲线(Curve)或微分热重曲线表示。
(2)影响热重测定的因素
1.升温速度
升温速度越快,温度滞后越大,Ti及Tf越高,反应温度区间也越宽。
对于无机材料试样,建议采用的升温速度一般为10-20K·min-1。
2.气氛
常见的气氛有空气、O2、N2、He、H2、CO2、Cl2和水蒸气等。
样品所处气氛的不同导致反应机理的不同。
气氛与样品发生反应,则TG曲线形状受到影响。
例如PP使用N2时,无氧化增重。
气氛为空气时,在150-180C出现氧化增重。
3.样品的粒度和用量
样品的粒度不宜太大、装填的紧密程度适中为好。
同批试验样品,每一样品的粒度和装填紧密程度要一致。
4.试样皿(坩锅)
试样皿的材质有玻璃、铝、陶瓷、石英、金属等,应注意试样皿对试样、中间产物和最终产物应是惰性的。
如聚四氟乙烯类试样不能用陶瓷、玻璃和石英类试样皿,因相互间会形成挥发性碳化物。
白金试样皿不适宜作含磷、硫或卤素的聚合物的试样皿,因白金对该类物质有加氢或脱氢活性。
5温度的标定
热天平可采用不同居里温度的强磁体来标定。
标定时在热天平外加一磁场,坩埚中放入标准磁性物质。
磁性物质的居里点是金属从铁磁性向顺磁性相转变的温度,在居里点产生表观失重。
三.示差扫描量热法DifferentialScanningCalorimeter,DSC
(1)差示扫描量热法(DSC)是在程序控温下,测量物质和参比物之间的能量差随温度变化关系的一种技术(国际标准ISO11357-1)。
根据测量方法的不同,又分为功率补偿型DSC和热流型DSC两种类型。
常用的功率补偿DSC是在程序控温下,使试样和参比物的温度相等,测量每单位时间输给两者的热能功率差与温度的关系的一种方法。
(2)与在DTA曲线中,吸热效应用谷来表示,放热效应用峰来表示所不同的是:
在DSC曲线中,吸热(endothermic)效应用凸起正向的峰表示凹下的谷表示(热焓增加),放热(exothermic)效应用凹下的谷表示(热焓减少)。
三.扫描电子显微镜(SEM)
透射电镜的成像——电子束穿过样品后获得样品衬度的信号(电子束强度),利用电磁透镜(三级)放大成像
扫描电镜成像——利用细聚焦电子束在样品表面扫描时激发出来的各种物理信号来调制成像的。
高能电子束与样品作用产生各种信息:
二次电子、背散射电子、吸收电子、特征X射线、俄歇电子、透射电子等
俄歇电子:
入射电子激发样品的特征X射线过程中,外层电子向内层跃迁时辐射出来的能量不是以X射线的形式发射出去,而被外层的另一个电子吸收而摆脱原子核的束缚而逃离出来,这个被电离的电子称为俄歇电子。
SEM的放大倍数为:
显像管中电子束在荧光屏上最大扫描幅度和镜筒中电子束在试样上最大扫描幅度的比值:
样品制备:
1..块状试样
对于块状导电材料,除了大小要适合仪器样品座尺寸外,基本上不需要进行什么制备,用导电胶把试样粘结在样品座上,即可放在扫描电镜中观察。
对于块状的非导电或导电性较差的材料,要先进行镀膜处理,在材料表面形成一层导电膜。
以避免电荷积累,影响图象质量,并可防止试样的热损伤。
2、粉末试样的制备
先将导电胶或双面胶纸粘结在样品座上,再均匀地把粉末样撒在上面,用洗耳球吹去未粘住的粉末,再镀上一层导电膜,即可上电镜观察。
3、镀膜
镀膜的方法有两种,一是真空镀膜,另一种是离子溅射镀膜。
通过加热蒸发某种物质使其沉积在固体表面,称为蒸发镀膜。
离子溅射镀膜的原理是:
在低气压系统中,气体分子在相隔一定距离的阳极和阴极之间的强电场作用下电离成正离子和电子,正离子飞向阴极,电子飞向阳极,二电极间形成辉光放电,在辉光放电过程中,具有一定动量的正离子撞击阴极,使阴极表面的原子被逐出,称为溅射,如果阴极表面为用来镀膜的材料(靶材),需要镀膜的样品放在作为阳极的样品台上,则被正离子轰击而溅射出来的靶材原子沉积在试样上,形成一定厚度的镀膜层
四.透射电镜
(1)TEM用聚焦电子束作照明源,使用于对电子束透明的薄膜试样,以透过试样的透射电子束或衍射电子束所形成的图像来分析试样内部的显微组织结构。
显微镜可分辨的两点间的最小距离,即为显微镜的分辨率
光学显微镜必须提供足够的放大倍数,把它能分辨的最小距离放大到人眼能分辨的程度。
相应的放大倍数叫做有效放大倍数
(2)透射电子显微镜样品制备
制样方法
a.粉末法b.化学减薄法c.双喷电解减薄法d.离子减薄法e.复型法
五.色谱
(1)色谱法定义、实质和目的
定义:
利用物质的物理化学性质建立的分离、分析方法
实质:
分离
目的:
定性分析或定量分析
(2)按分离机制分
分配色谱:
利用分配系数的不同
分离机制:
各组分与流动相分子争夺吸附剂表面活性中心
利用吸附剂对不同组分的吸附能力差异而实现分离
吸附色谱:
利用物理吸附性能的差异
分离机制:
利用组分在流动相和固定相间溶解度差别实现分离离子交换色谱:
利用离子交换原理
分离机制:
依据被测组分与离子交换剂交换能力(亲和力)
不同而实现分离
空间排阻色谱:
利用排阻作用力的不同
分离机制:
利用被测组分分子大小不同、在固定相上选择性
渗透实现分离
(3)色谱分离原理
色谱分离基于各组分在两相之间平衡分配的差异,平衡分配可以用分配系数和分配比来衡量
(4)色谱分离特点
1.不同组分通过色谱柱时的迁移速度不等→提供了分离的可能性
2.各组分沿柱子扩散分布→峰宽↑→不利于不同组分分离
六.红外光谱
(1)红外光谱是一种振动光谱;
当光和物质相互作用时,会引起物质分子或原子基团的振动,从而产生对光的吸收,这样产生的光谱称为振动光谱。
如果用的光源是在红外光的范围(0.75-1000µm),就是红外吸收光谱;如果用的是强单色光(如激光),产生的是激光拉曼光谱。
(2)光的二重性
光同时具有波动性和粒子性。
它既是一种振动波,又是一种以高速度移动的粒子流,可以把它称为光量子或光子。
在表示光的特征时,
波的基本公式:
c=λν(波动性)
普朗克公式:
E=hν(粒子性)
2.多原子分子的振动——简正振动
①振动自由度
由N个原子构成的复杂分子的原子振动有多种形式,通常称为多原子分子的简正振动。
多原子分子简正振动的数目称为振动自由度。
多原子分子振动比双原子要复杂得多。
要描述多原子分子各种可能的振动方式,必须跟定各原子的相对位置。
在含有N个原子分子中,其位置可用简正坐标描写,而每个原子的运动可以用三个位移坐标来表达。
因此该分子被认为有3N个自由度。
N个原子是由化学键构成的一个整体分子,因此必须从分子整体来考虑自由度。
分子作为整体有三个平动自由度和三个转动自由度,剩下(3N-6)才是分子的振动自由度(直线型分子有3N-5个振动自由度)。
每个振动自由度相应于一个基本振动,N个原子组成的非线形分子,共有3N-6个基本振动,这些基本振动称为分子的简正振动。
对于红外光谱法来说,要产生振动吸收需要有两个条件,即:
分子振动的频率与红外光谱中某段频率相同,或者说,IR光的某些光子能量要与分子振动能相吻合。
——必要条件
振动必须引起偶极矩变化,才是IR活性的。
即正负电荷中心的间距发生变化。
——IR活性振动。
分子类型(偶极矩)与红外活性的一般关系:
对于双原子分子:
①若为极性分子,则它具有偶极矩,为红外活性,如HX、CHCl3等存在红外光谱;
②若为非极性分子,则它无偶极矩,为非红外活性,如N2、O2、Cl2等无红外吸收光谱。
对于多原子分子:
视具体情况来判断是否有偶极矩,从而确定是否具有红外活性。
如CO2虽然为非极性分子,但它在2368、668cm-1存在
红外吸收峰。
红外光谱图的定义及表示方法
⏹定义:
用来研究波数在4000-400cm-1范围内不
同波长的红外光通过化合物后被吸收的谱图
红外光谱图的特征(四个要素)
谱带的数目、谱带的位置、谱带的形状、谱带的强度
红外光谱图的影响因素
①影响峰位变化的因素
②影响峰强的因素
A、偶极矩的变化B、能级的跃迁几率
官能团区(4000~1300cm-1):
即化学键和基团的特征振动频率部分,主要反映分子中特征基团的振动,基团的鉴定工作主要在这一光谱区域进行。
有机物的基团振动主要占据官能团区,无机物较少
指纹区(1300~400cm-1):
比较复杂,反映分子结构的细微变化。
每一种化合物在该区的谱带位置、强度和形状都不一样,相当于人的指纹,因而称之为指纹区。
指纹区也有一些特征吸收带,可用于鉴定官能团。
无机物的基团振动主要占据指纹区。
红外光谱的样品制备方法
1、固体样品的制备
①卤化物压片法②粉末法③糊状法④薄膜法
2、液体和气体样品的制备
①液体样品
溶液法:
把液体注入液体槽中进行测定。
溶剂常用CCl4或CS2,液体槽有可拆卸式和固定密封式两种。
液膜法:
在两个窗片之间,滴上1~2滴液体试样,形成一层薄的液膜用于测定。
此方法只适用于高沸点液体化合物。
②气体样品
一般使用气体槽进行测定。
七.紫外光谱
电子跃迁类型:
1.σ→σ*跃迁:
饱和烃(甲烷,乙烷)
E很高,λ<150nm(远紫外区)
2.n→σ*跃迁:
含杂原子饱和基团(—OH,—NH2)
E较大,λ150~250nm(真空紫外区)
3.π→π*跃迁:
不饱和基团(—C=C—,—C=O)
E较小,λ~200nm
体系共轭,E更小,λ更大
4.n→π*跃迁:
含杂原子不饱和基团(—C≡N,C=O)
E最小,λ200~400nm(近紫外区)
饱和烃(甲烷,乙烷)
E很高,λ<150nm(远紫外区)
紫外光谱电子跃迁类型:
n—π*跃迁π—π*跃迁
饱和化合物无紫外吸收
生色团(发色团):
能吸收紫外-可见光的基团
有机化合物:
具有不饱和键和未成对电子的基团
具n电子和π电子的基团
产生n→π*跃迁和π→π*跃迁
跃迁E较低
例:
C=C;C=O;C=N;—N=N—
助色团:
本身无紫外吸收,但可以使生色团吸收峰加强同时使吸收峰长移的基团
有机物:
连有杂原子的饱和基团
例:
—OH,—OR,—NH—,—NR2—,—X
红移和蓝移:
由于化合物结构变化(共轭、引入助色团取代基)或采用不同溶剂后
吸收峰位置向长波方向的移动,叫红移(长移)
吸收峰位置向短波方向移动,叫蓝移(紫移,短移)
Ø影响吸收带位置的因素:
1.溶剂效应:
Ø对λmax影响:
next
n-π*跃迁:
溶剂极性↑,λmax↓蓝移
π-π*跃迁:
溶剂极性↑,λmax↑红移
Ø对吸收光谱精细结构影响next
溶剂极性↑,苯环精细结构消失
✓溶剂的选择——极性;纯度高;截止波长<λmax
2.pH值的影响:
影响物质存在型体,影响吸收波长
八.拉曼光谱(Raman光谱)
当光照射到物质上时会发生非弹性散射,散射光中除有与激发光(入射光)波长相同的弹性成分(瑞利散射)外,还有比激发光波长长的和短的非弹性成分统称为拉曼散射。
由分子振动、固体中的光学声子等元激发与激发光相互作用产生的非弹性散射称为拉曼散射。
一般把瑞利散射和拉曼散射合起来所形成的光谱称为拉曼光谱。
拉曼光谱为40–4000cm-1,而红外光谱测试一般位于中红外区(400–4000cm-1)。
九.能谱
1、X射线光电子能谱
(X-rayphotoelectronspectroscopy,XPS)
2、俄歇电子能谱(Augerelectronspectroscopy,AES)
它是用具有一定能量的电子束(或X射线)激发样品俄歇效应,通过检测俄歇电子的能量和强度,从而获得有关表面层化学成分和结构的信息的方法。
俄歇过程至少有两个能级和三个电子参与,所以氢原子和氦原子不能产生俄歇电子。
光电效应图:
光电效应(光电吸收)
当X射线的波长足够短时,其光子的能量就很大,以至能把原子中处于某一能级上的电子打出来,而光子本身则被吸收;它的能量就传递给该电子,使之成为具有一定能量的光电子,并使原子处于高能的激发态。
这种以光子激发原子所发生的激发与辐射的过程称为光电吸收或光电效应。
X射线衍射分析:
XRD
X射线0.01~10nm
布拉格公式:
●θ:
入射线(或反射线)与晶面的夹角;也称
为布拉格角、半衍射角或掠射角;
●2θ:
入射线与衍射线之间的夹角;衍射角。
●布拉格方程是X射线晶体学中最基本的公式,是衍射
中最基本、最重要的方程。
●布拉格方程表明,要形成衍射,d、θ、λ必须满足
布拉格方程。
●与光学反射定律加在一起,就是布拉格定律。
布拉格证实在7大晶系中,只可能有
14种布拉格点阵。
●7大晶系—4种空间点阵(每个晶系最多)
14种布拉格点阵(所有晶系实际)。
7个晶系:
14种布拉菲点阵
立方晶系:
P、I、F;
正方晶系:
P、I;
斜方晶系:
P、I、F、C;
菱方晶系:
P;
六方晶系:
P;
单斜晶系:
P、C;
三斜晶系:
P。
X射线衍射的几何条件(衍射方向)
X射线照射晶体,电子受迫振动产生相干散射;同一原子内各电子散射波相互干涉形成原子散射波。
由于晶体内各原子呈周期排列,因而各原子散射波间也存在固定的位相关系而产生干涉作用,在某些方向上
发生相互干涉,即形成衍射波。
由此可知,衍射的本质是晶体中各原子相干散射波叠加(合成)的结果。
衍射波的两个基本特征:
衍射线在空间分布的方位(衍射方向)和强度。
能谱
能谱仪全称为能量分散谱仪(EnergyDispersiveX-raySpectrometer,EDS,EDX).
功能:
化学元素定性、定量和分布影像分析
每种元素具有自己特定的x射线特征波长,而特征波长的大小则取决于能级跃迁过程中释放出的特征能量△E。
能谱仪就是利用不同元素x射线光子特征能量不同这一特点来进行成分分析的,其强度决定元素的含量。
△E=hν=hc/
 升级会员
升级会员 