Gauss计算步骤.docx
《Gauss计算步骤.docx》由会员分享,可在线阅读,更多相关《Gauss计算步骤.docx(7页珍藏版)》请在冰点文库上搜索。
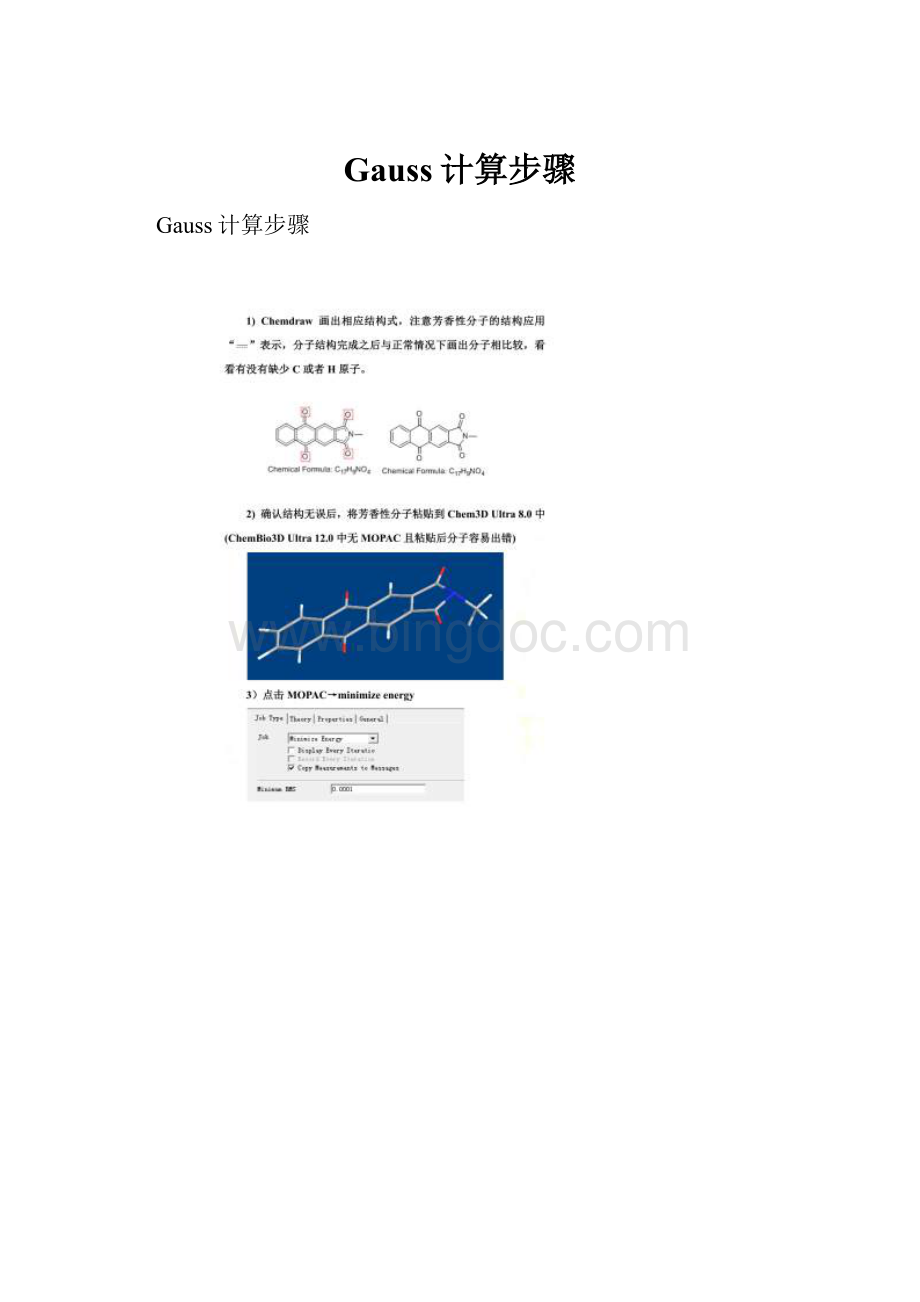
Gauss计算步骤
Gauss计算步骤
此处计算的是自由基负离子所以只能选择openshell,unrestrictedopen
shell表示分别计算α和β电子,自由基阴离子net=-1,spin为2,自旋多重度=2S+1,即单电子数目加1
6)点击create之后弹出保存按钮对话框,保存成.gjf文件
7)启动GaussView5.0,file→open打开刚才保存的.gjf文件
8)calculate→Gaussiancalculationsetup
此处选择optimization,收敛标准选择usetightconvergencecriteria
Method选择基态;charge=-1,spin=2;选择极化函数+弥散函数
Jobtitle用于给文件命名
此处memorylimit用于指定计算所占内存大小,高斯使用默认的内存48M,1MW=8MB;sharedprocessors用于指定多核处理器,本实验室计算工作站可以选择4,高斯默认为1.
一般进行结构优化以及频率计算的时候不保存chkpointfile文件,等单点能以及激发态计算的时候才保存chkpointfile文件。
一般ignoresymmetry,即忽略分子对称性,因为有时计算结果分子对称性会发生变化;UseMaxdisk用于指定计算所用硬盘大小,默认2G,一般都设置的比较大一点,要不然计算可能出错,我一般用10G或者20G.再后面的Guess(初始猜测)、NBO(成键轨道分析)和Solvation(溶剂化)不用管。
9)参数设置完成,在检查一遍无错误后,点击edit→save即保存Gaussianoptimization的input文件,文件类型Gaussianinputfile。
10)点击save会弹出一个写字板文件信息,上面分别记载了计算的一些设置以及原子坐标信息
11)关闭写字板文件,会弹出一个提交Gaussian计算的对话框,点击cancel。
12)启动Gaussian09,file→open打开刚才保存的Gaussianinputfile文件,再次确认计算指令信息无误后点击软件右上角的run按钮,即可进入计算程序进行构象优化。
13)计算结束,得到高斯计算结果GaussianOutputFile,构象优化是计算最耗时部分,一般大于10个小时。
14)打开Gaussview,file→open打开构象优化得到的GaussianOutputFile文件,calculate→Gaussiancalculationsetup进行下一步频率计算的参数设置。
Jobtype选择frequency,其他选择默认不进行改动,注意频率计算必须和构象优化使用同一基组,要不然频率计算将失去意义。
15)同构象优化一样,参数设置完毕,点击edit,保存文件为Gaussianinputfile,按照构象优化相同程序进入频率计算。
16)频率计算结束后,输出文件是GaussianOutputFile,打开Gaussview,file→open打开频率计算得到的GaussianOutputFile文件,点击Results→vibration,弹出如下对话框,检查有无虚频:
无虚频则说明得到的是最有构象,出现虚频则说明构象优化不正确,可以改变初始给系统的构象、分子对称性、收敛标准等重新优化。
OPT(MAXCYCLE=200,MAXSTEP=1)
1Hartree=627.5kcal/mol=27.21138ev
 升级会员
升级会员 