药事管理学部分考试知识点文档格式.doc
《药事管理学部分考试知识点文档格式.doc》由会员分享,可在线阅读,更多相关《药事管理学部分考试知识点文档格式.doc(4页珍藏版)》请在冰点文库上搜索。
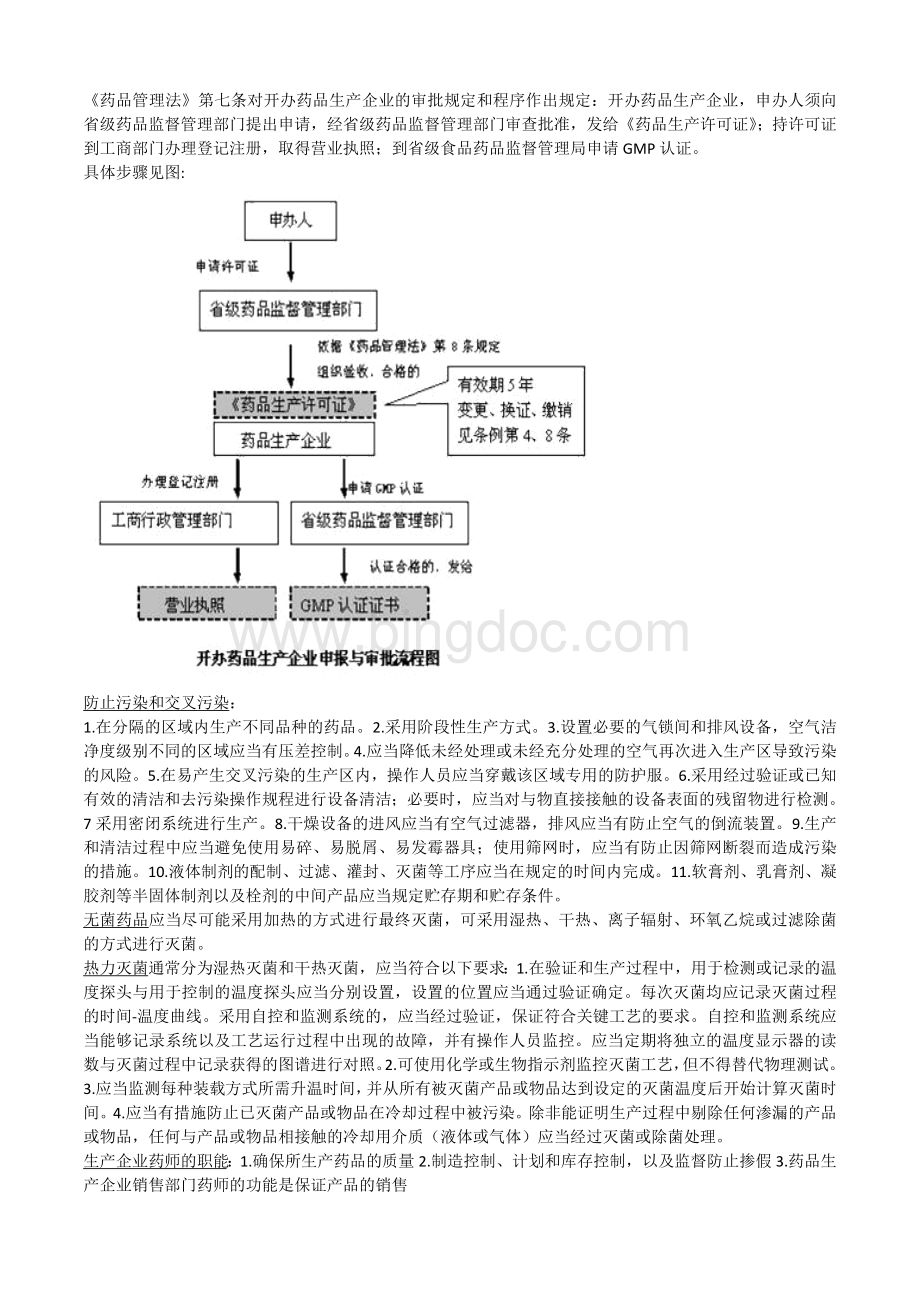
使用筛网时,应当有防止因筛网断裂而造成污染的措施。
10.液体制剂的配制、过滤、灌封、灭菌等工序应当在规定的时间内完成。
11.软膏剂、乳膏剂、凝胶剂等半固体制剂以及栓剂的中间产品应当规定贮存期和贮存条件。
无菌药品应当尽可能采用加热的方式进行最终灭菌,可采用湿热、干热、离子辐射、环氧乙烷或过滤除菌的方式进行灭菌。
热力灭菌通常分为湿热灭菌和干热灭菌,应当符合以下要求:
1.在验证和生产过程中,用于检测或记录的温度探头与用于控制的温度探头应当分别设置,设置的位置应当通过验证确定。
每次灭菌均应记录灭菌过程的时间-温度曲线。
采用自控和监测系统的,应当经过验证,保证符合关键工艺的要求。
自控和监测系统应当能够记录系统以及工艺运行过程中出现的故障,并有操作人员监控。
应当定期将独立的温度显示器的读数与灭菌过程中记录获得的图谱进行对照。
2.可使用化学或生物指示剂监控灭菌工艺,但不得替代物理测试。
3.应当监测每种装载方式所需升温时间,并从所有被灭菌产品或物品达到设定的灭菌温度后开始计算灭菌时间。
4.应当有措施防止已灭菌产品或物品在冷却过程中被污染。
除非能证明生产过程中剔除任何渗漏的产品或物品,任何与产品或物品相接触的冷却用介质(液体或气体)应当经过灭菌或除菌处理。
生产企业药师的职能:
1.确保所生产药品的质量2.制造控制、计划和库存控制,以及监督防止掺假3.药品生产企业销售部门药师的功能是保证产品的销售
有下列情况之一的为假药:
1.药品所含成分与国家药品标准规定的成分不符的;
2.以非药品冒充药品或者以其他种药品冒充此种药品的。
有下列情形之一的药品,按假药论处:
国务院药品监督管理部门规定禁止使用的;
2.依照本法必须批准而未经批准生产、进口或者依照本法必须检验而未经检验即销售的;
3.变质的;
4.被污染的5.使用依照本法必须取得批准文号而未取得批准文号的原料生产的;
所标明的适应症或者功能主治超出规定范围的。
劣药指药品成分的含量不符合国家药品标准的。
有下列情形之一的药品,按劣药论处:
1.未标明有效期或者更改有效期的;
2.不注明或更改生产批号的;
3.超过有效期的;
4.直接接触药品的包装材料和容器未经批准的;
5.擅自添加着色剂、防腐剂、香料、矫味剂及辅料的;
6.其他不符合药品标准规定的。
仿制药申报与审批程序与新药相似,即由省级药品监督管理部门受理并进行形式审查和研制(临床试验)现场核查,药品审批中心进行技术审核并提出技术审评意见,最终由国家食品药品监督管理局审批。
不同的是仿制药的药品生产现场检查主要由省级药品监督管理部门负责。
完成临床试验者,报送临床试验资料
不同意
检验报告
《药品注册申请表》,申报资料
样品
申请人
省级药品监督管理局
指定的药检所
注册检验
药品注册批件
药品批准文号
同意
需要临床试验者,发给《药物临床试验批件》
审批意见通知件
抽取连续3批号样品,对生产条件现场考查,资料形式审查
药品审评中心
国家食品药品监督管理局
审评
新药注册特殊审批的情形:
a.未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂,新发现的药材及其制剂;
b.未在国内外获准上市的化学原料药及其制剂、生物制品;
c.治疗艾滋病、恶性肿瘤、罕见病等疾病且具有明显临床治疗优势的新药;
d.治疗尚无有效治疗手段的疾病的新药。
其中主治病症状未在国家批准的中成药“功能主治”中收载的新药,可以视为尚无有效治疗手段的疾病的新药。
药品广告有关药品功能疗效的禁止性规定:
①含有不科学地表示功效的断言或者保证的。
②说明治愈率或者有效率的。
③与其他药品的功效和安全性进行比较的。
④违反科学规律,明示或者暗示包治百病、适应所有症状的。
⑤含有“安全无毒副作用”、“毒副作用小”等内容的;
含有明示或者暗示中成药为“天然”药品,因而安全性有保证等内容的。
⑥含有明示或者暗示该药品为正常生活和治疗病症所必需等内容的。
⑦含有明示或暗示服用该药能应付现代紧张生活和升学、考试等需要,能够帮助提高成绩、使精力旺盛、增强竞争力、增高、益智等内容的。
⑧其他不科学的用语或者表示,如“最新技术”、“最高科学”、“最先进制法”等。
SFDA国家食品药品监督管理局。
药品不良反应英文(adversedrugreaction,ADR)。
ADR发生率的表示方法:
十分常见:
≥1/10;
常见:
≥1/100且<
1/10;
偶见:
≥1/1000且<
1/100;
罕见:
≥1/10000且<
1/1000;
十分罕见:
<
1/10000。
A、B、C型ADR三类。
药物临床试验:
临床试验和生物等效性试验。
临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。
临床试验的最低受试者:
Ⅰ期为20~30例,II期为100例,III期为300例,IV期为2000例。
预防用生物制品的临床试验的最低受试者:
Ⅰ期为20例,II期为300例,III期为500例。
麻醉药品按药理作用:
镇痛类、非镇痛类。
精神药品按药理作用:
镇静催眠类、中枢兴奋类、镇痛及复方制剂类、全身麻醉药等.
第一类精神药品和第二类精神药品
中药二级保护:
7年
处方药RX。
非处方药OTC,甲类:
红底白字;
乙类:
绿底白字。
药物研发:
临床前研究(preclinicalstudy)和临床研究(clinicalstudy)
药品检验机构:
(一)中国食品药品检定研究院(NIFDC)
(二)省、自治区、直辖市药品检验所.
中国食品药品检定研究院是最高技术仲裁机构
药品批准文号的格式为:
国药准字H(Z、S、J)+4位年号+4位顺序号,其中H代表化学药品,Z代表中药,S代表生物制品,J代表进口药品分包装。
药品生产的特点:
1.产品的种类和规格多、消耗大
2.机械化、自动化程度要求高
3.生产过程卫生要求严格
4.产品质量基线要求高
5.生产质量管理法制化
《药物非临床研究质量管理规范》GoodLaboratoryPracticeGLP
《药物临床试验质量管理规范》GoodClinicalPracticeGCP
《药品生产质量管理规范》GoodManufacturingPracticeGMP
《药品经营质量管理规范》GoodSupplyPracticeGSP
药学职业道德原则:
保证药品质量,保障人体用药安全,维护人们用药的合法权益,实行社会主义,人道主义,全心全意为人民身心健康服务。
具体原则为:
质量第一的原则,不伤害原则,公正原则,尊重原则。
药品标签书写印制要求:
药品通用名称应当显著、突出,其字体、字号和颜色必须一致,并符合以下要求:
①对于横版标签:
必须在上1/3范围内显著位置标出;
对于竖版标签:
必须在右1/3范围内显著位置标出。
②不得选用草书、篆书等不易识别的字体,不得使用斜体、中空、阴影等形式对字体进行修饰。
③字体颜色应当使用黑色或者白色,与相应的浅色或者深色背景形成强烈反差。
④除因包装尺寸的限制而无法同行书写的,不得分行书写。
要求申请人补充资料
技术审评
抽样
新药申请人
省级药品监督管理部门
填写《药品注册申请表》,报送资料
形式审查、现场核查
初审
国家局药品审评中心
药品检验所
审查意见、核查报告,申报资料
国家局审批
技术审评意见
相关资料
《药物临床试验批件》
《审批意见通知书》
新药生产申请与审批流程图
 升级会员
升级会员 