生化线性分析.docx
《生化线性分析.docx》由会员分享,可在线阅读,更多相关《生化线性分析.docx(46页珍藏版)》请在冰点文库上搜索。
前面写过“生化分析仪维修有关的基础知识”(
1、导向知识:
生化分析仪的基本原理是分光光度计,或者俗称比色计。
分光光度计的依据是“朗伯—比尔定律”。
朗伯—比尔定律阐述了液体吸光度与液体浓度的关系,并且引申出相应的公式及推导公式。
吸光度越高,溶液的色度也就越深,反之越浅。
当然前提是同波长下。
一般来说,生化反应把吸光度增加的叫做正反应,或者叫做上升反应,色度越来越深;吸光度下降的反应叫做负反应或者下降反应,色度越来越浅。
应用和维修的界限其实很难划分,一般来说操作问题属于应用,故障属于维修。
但结果问题有可能是应用问题,也有可能是故障,所以生化仪区分应用和维修我认为纯属找麻烦。
2、生化的测试方法:
从分光光度计的方法来说,有透射和散射两种方法,生化仪只用到透射法,因为它只有一套光路。
贝克曼的自有机型和特定蛋白仪及免疫类血凝类设备,还增加有散射法等等。
生化的测试方法只有两种,那就是终点法和速率法,其余方法都是衍生法。
而单试剂或者双试剂与否与方法关系不大,只跟衍生法有关。
不过单试剂的大多是终点法,没见过有单试剂速率法的。
2.1 终点法顾名思义,在反应终点进行吸光度测定的方法,其衍生方法有一点终点法,对应单试剂;两点终点法对应双试剂。
还有一些相关的概念:
试剂空白、血清空白。
先声明一下,下面出现的所有例图都是选自日立、奥林巴斯、东芝、拜耳这些生化仪的手册,选择的目的一是有代表性,而是清晰度好,并非我个人有所倾向。
2.2 一点终点法:
也就是单试剂采用的方法。
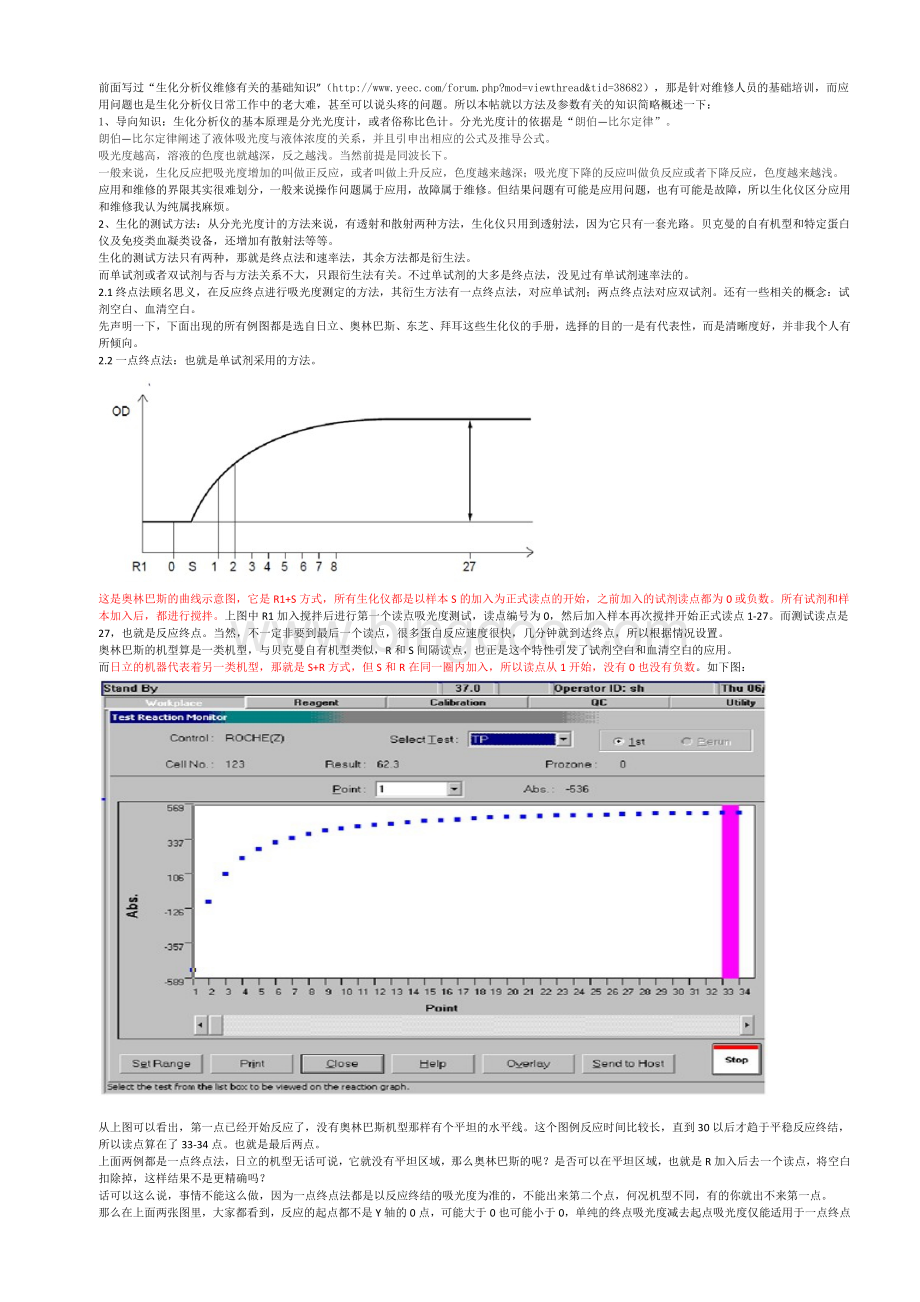
这是奥林巴斯的曲线示意图,它是R1+S方式,所有生化仪都是以样本S的加入为正式读点的开始,之前加入的试剂读点都为0或负数。
所有试剂和样本加入后,都进行搅拌。
上图中R1加入搅拌后进行第一个读点吸光度测试,读点编号为0,然后加入样本再次搅拌开始正式读点1-27。
而测试读点是27,也就是反应终点。
当然,不一定非要到最后一个读点,很多蛋白反应速度很快,几分钟就到达终点,所以根据情况设置。
奥林巴斯的机型算是一类机型,与贝克曼自有机型类似,R和S间隔读点,也正是这个特性引发了试剂空白和血清空白的应用。
而日立的机器代表着另一类机型,那就是S+R方式,但S和R在同一圈内加入,所以读点从1开始,没有0也没有负数。
如下图:
从上图可以看出,第一点已经开始反应了,没有奥林巴斯机型那样有个平坦的水平线。
这个图例反应时间比较长,直到30以后才趋于平稳反应终结,所以读点算在了33-34点。
也就是最后两点。
上面两例都是一点终点法,日立的机型无话可说,它就没有平坦区域,那么奥林巴斯的呢?
是否可以在平坦区域,也就是R加入后去一个读点,将空白扣除掉,这样结果不是更精确吗?
话可以这么说,事情不能这么做,因为一点终点法都是以反应终结的吸光度为准的,不能出来第二个点,何况机型不同,有的你就出不来第一点。
那么在上面两张图里,大家都看到,反应的起点都不是Y轴的0点,可能大于0也可能小于0,单纯的终点吸光度减去起点吸光度仅能适用于一点终点法。
所以奥林巴斯机型把R加入后的平坦区域,叫做试剂空白,这没错,这段区域只有R参与,根本没有任何样本。
但它并不在测试时增加一个空白读点,而是在测试准备或定标准备前增加了一个试剂空白测试。
试剂空白检测完毕后,数据保存在数据库里,在更换试剂之前,试剂到期之前都采用这个数据作为空白值参与结果的计算。
试剂空白有一定的范围,超过这个设定范围会认为试剂失效或者污染,所以在奥林巴斯的机型里,试剂空白是一定要做的,而且间隔时间不能太长,否则会报错。
除了试剂空白外,还有血清空白,因为血清的底色甚至高于试剂底色。
所以在一点终点法中,为了得到精确的结果,也要进行血清空白的测试。
要注意的是,这个方法很少使用,因为要测定血清空白需要单独的通道。
也就是说一个样本要被测试两次,一次是常规的反应,加入R进行测试,另一次是单独采集样本,不加入试剂,而加入生理盐水或纯水稀释到前一次的稀释比例,单独测试样本血清的吸光度。
然后将终点反应的吸光度减去血清空白,得到的结果才是反应结果。
如下图:
2.3 两点终点法,也就是双试剂常用的终点法。
在奥林巴斯机型中,双试剂会出现两个或三个平坦的区域,R1加入之后,和S加入之后,如果是终点法,会在反应终点还会出现一个。
如下图:
那么两点终点法除了在27点读一个之外,还要在10点读一个点,也就是在R2加入之前读一个点。
这样反应吸光度值就等于终点吸光度值减去试剂样本空白,注意是试剂样本空白,不是试剂空白,所以这个点只能在S之后,R2之前。
惯例一般在R2之前的一个点,而不是R2之前S之后的任意点。
下面是日立的图例:
由于是S+R1,所以曲线开始就进行反应,除去杂质消除干扰,直到R2加入前趋于稳定。
16点之后加入R2,正式反应启动,直到27点趋于稳定,所以终点读点选在33-34上。
而试剂样本空白选择在15-16上。
从日立的图上我们可以看出,如果选择一点终点法,那么反应的吸光度值应该是137-(-562)=699,而去除试剂样本空白的两点终点法的反应吸光度值呢?
应该是137-(-477)=614,可以看出后者更为精确。
说明:
如果是正反应,吸光度值等于终点值-试剂样本空白值;如果是负反应,吸光度值等于终点值-(-试剂样本空白值)。
这就是为什么要告诉反应方向的原因,否则算出来是负的结果没法报了。
总结:
一点终点法适用于单试剂,两点终点法适用于双试剂,但第一点要在R2之前的一点。
2.4 速率法:
使用最小平方法计算两点间的吸光度变化,确定每分钟吸光度的变化率,也就是ΔOD/min。
常规的速率法是在R2加入之后,给出两个读点,系统根据两点间的时间,自动计算每分钟的吸光度变化。
下图是奥林巴斯的图例:
下图是日立的图例:
可以看出上图读点范围是22-28点。
采用常规速率法有个前提,那就是R1加入之后前期反应趋于稳定,比较平坦,加入R2之后迅速启动反应,形成斜线,读点范围取在斜线上。
如果R1加入之后也是斜线,那就要使用双速率法,也就是在试剂样本空白的斜线上取一段进行空白速率计算。
双速率法很少使用,与常规速率法的结果比较差别也不是很大。
在日立机型中,常规速率法叫速率A法。
日立多了一个速率B法,那是双项目检测的,没有专用试剂无法实现。
图例中的速率法每点线性都很好,但遇到每点线性不好的,就会报错,结果被拒绝、屏蔽。
线性不好一方面是试剂质量问题,另一方面是反应特征,无法保持线性。
那么这种问题各个厂家也都有对应,日立叫做两点速率法,奥林巴斯叫做固定点法。
2.5两点速率(固定点)法:
确定特定两点间的吸光度差值,无线性甄别。
还有引申出来的双固定点法,对应双速率法:
上面两图都是奥林巴斯的,下面是日立的两点速率法。
2.6终点法和速率法的运用不能混淆,什么项目用什么方法一般不会有错,试剂厂家的说明书上都有明确介绍,一般不会搞错,但也有例外,下图就是一个例子,是小东版主的讲义节选的。
例1:
故障现象是TBA结果总是出现负值,而且更换过试剂,定标都通过,仍然解决不了。
曲线如下:
方法采用的是两点终点法(R2之前之后各一点),这是致命的错误。
根本找不到终点的曲线,怎么能使用终点法呢?
再来看看负值是怎么来的,TBA是正反应,如果采用两点终点法应该是后点-前点。
左图后点高于前点,所以是正值;而右图后点低于前点,减出来肯定是负值。
但方法错误这个前提已经确定,后面再怎么分析也没有意义了,明显的速率法。
读点问题最常见,这也就是结果问题要找曲线的原因所在。
例2:
这是群里的一个问题:
高低密项目定标和质控都没有问题,就是病人结果偏低。
这个无法直接回答,还是上曲线看看吧。
两张图传完就明白原因了,读点错误。
高低密采用两点终点法,而两点终点法的关键是R2之前一点,另一点在反应的终点。
而图上明显是两点都在R2之后,再分析为什么会结果偏低,以第一张图为例。
这个机器是R1+S方式,R1加入后吸光度在400上下,比较平稳,紧接着加入S,吸光度升到1000左右,加入R2之后,反应终点区域大致在1300左右。
如果是R2之前一点为1000,R2之后反应终点为1300,那么两点终点法的反应吸光度为1300-1000=300,据此计算浓度。
而图例中两点都选择在了R2之后,第一点的吸光度大致在1300左右,第二点也是1300左右,二者相减相差无几,搞不好还是负的,难怪结果低了。
有一点我很不理解,生化仪的软件设计人性化方面有问题。
既然选择了终点法,你就不该让操作人员设置读点的时候把第一点设为R2之后,干脆报错提示多好呢?
2.7速率法的底物过剩问题。
所谓底物过剩,就是样本浓度太高,试剂提供的反应物质迅速反应完,而样本中的检测物质还有很多没有参与反应。
通俗的话讲,就是在速率法的反应中看到了终点法的曲线。
下面是图例,也是小东版主的讲义:
图例中看到的是典型的日立速率A法,见到这样的图,结果肯定偏低甚至为0或者负值。
这样的图首先应该想到方法是不是错误,如果速率法没错,那就只有一个可能,底物过剩。
几乎所有的机型都提供危急值报警和自动重测,遇到这种情况,一般都采取自动或人工稀释重测的方法解决。
而东芝及其OEM机型雅培的生化仪,还有日电及拜耳的机型,可以自动进行判别,并将读点前移。
东芝叫做弹性速率法,日电叫做读点前移。
概念是这样的:
以上右图为例,速率法选择的主读点区为22-28,并加以吸光度判别和线性判别,当出现底物过剩时,这个主读点区的线性几乎停滞,吸光度变化降低到几乎为0,这种情况下启动弹性速率读点区,一般设置在R2加入后10点以内,比如选择在17-21这个区间里。
在弹性速率区间里进行速率法计算,从而得出一个较高的浓度值,因为这一区间反应速度很快。
这种方法以前有很多医院的老师作过实验并发表过论文,与手工稀释的结果比对,符合率高达96%。
这样一来,省工省力,节省了医院最为看重的成本。
下图是官方的弹性速率解释和读点前移介绍:
总结:
终点法记住关键的R2前的一点,速率法的读点都在R2之后,两点速率法或固定点法不检查线性,底物过剩要看曲线。
3、生化的定标方法:
定标方法分为线性定标和非线性定标,也就是俗话说的直线定标和曲线定标。
生化中大部分项目都是用线性定标,而非线性定标大多用在免疫比浊项目上。
3.1线性定标。
我们都知道,两点决定一线,所以要定标最少要有2个定标点。
定标的概念是在直角坐标系中,以浓度为X轴,吸光度值为Y轴,通过校准点的XY轴对应关系,将直线确定下来,标本测试中所有的吸光度值都能在这条直线上找到对应的浓度值。
如下图所示:
图中给出的STD1、2两点是定标液,无论它们的吸光度值是多少,都会有一个确定的浓度值赋于它们,那么在直角坐标系中,这条定标曲线就被确定下来,无论样本的吸光度值是多少,都能在这条线上找到对应的浓度值。
同时我们还能看出,这条直线是这个第一象限的角平分线,也就是45度的倾角。
这种定标的结果保证了很宽的线性范围,无论多高多低都会有良好的线性。
如果这个倾角大于或小于45度,那么在宽泛的测试中,吸光度的变化与浓度的对应就不能好计算了。
所以很多高值或低值结果不好,而且又是线性定标的话,就要看看定标曲线是不是45度的倾角,如果不是,就要考虑定标问题了。
那么定标的直线是怎么来的呢?
根据直线方程而来,也就是一元一次方程,Y=AX+B。
Y是吸光度值,X是浓度值,A是因数,B是截距。
因数也被称为斜率、K值。
再通俗的讲,A其实就是这条直线的倾角,B是X=0的值,也就是直线与Y轴的交点。
所以得到这条直线的方法有三种:
I、假设直线过原点,也就是B=0,那么只要给出A的值,直线也就可以得出。
当然如果要想得到45度的漂亮曲线,至于要将A=1即可,也就是Y=X。
这个A大部分时候要乘以10000,这是一个换算因素。
也就是很多厂家给出的K值是1万多,8千多等等的原因。
这就是常说的K因数线性定标法。
II、假设直线过原点,也就是B=0,那么只要有一个非零点的值,就可以得到整条值线。
因为第一个值已经确定,那就是Y和X都是0,也就是原点。
这就是常说的一点线性定标。
III、给出两个定标点,让机器自己计算直线是否过原点,这就是两点线性定标。
我们可以看出三种方法前两种都有一个前提,那就是假设直线过原点。
那么到底这个项目是否过原点呢?
我们并不肯定,所以I、II两种方法都有缺陷,不太靠谱。
特别是I种方法,无法得知厂家给出的K值是怎么来的,如何界定准确与否。
下图是因数K值定标和假设过原点的一点线性定标示意图:
所以推荐的方法,也是不出错的,最为准确的方法是III,两点线性定标。
在两点线性定标中,大多数时候是给一个标准点和水点,水点的意义是给一个零浓度的点。
这样一个零浓度,一个有浓度,两个点决定一条直线。
当然,零浓度点也可以用带有浓度值的标准替代,无论如何只要给两点就能决定一条直线。
下图就是示意图:
K因数法定标虽然不靠谱,但大多数医院特别是小医院还是喜欢用,因为毫无成本,不需要校准品。
但也正是因为这样的不靠谱,导致所有采用这种方法校准的项目,结果往往出现高值不高,低值不低,甚至与其它机器无法做出准确的对比。
假设过原点的定标又叫一点线性定标,只需要一个校准品即可。
但有一个问题,你确信它绝对过原点?
如果不过原点,那么就存在一个截距,结果总是差那么多,又不能改系数,非常别扭。
在日立机型上,三种线性定标的方法分别称为1点定标(K因数法),两点定标。
东芝和拜耳与之类似。
在奥林巴斯的机器上ACALAA法针对的是两点线性定标,给出两个浓度点(其中一个可以是零浓度)自动进行直线矫正,自动判断是否过原点。
而ACALAB则是假定过原点,再给出一个浓度点即可,也就是一点线性定标。
MCALMB法则是K因数法。
奥林巴斯经常出现结果不准的问题,很大程度上与定标方法的选择有关,大部分都选择ACALAB或者干脆使用MCALMB。
抛开K因数法的MCALMB,单说ACALAB。
其实我们无法得知这个项目或者这个试剂是否真的过原点,是否总是过原点,因此ACALAA是最佳的选择,因为并不增加成本,只是给一个水点,也就是零浓度点罢了。
由于奥林巴斯机型软硬件设计上的特点,把简单的定标搞的云山雾罩的,特别是几种颜色的架子,把人们搞糊涂了。
而且很多人把试剂空白和水点混为一谈,造成定标方面的诸多问题。
试剂空白是奥林巴斯机型必须要做的,为了保证结果的准确,在测试吸光度上减去试剂空白是正确的。
而试剂空白并不是零浓度点,两个完全不同的概念。
试剂空白是各个测试都要参与的,零浓度点只在定标时使用。
虽然二者使用的样本都是水。
奥林巴斯机型中,蓝色架子是试剂空白,这个在测试前要先放入进样器,紧跟着是定标用的黄色架子。
我们一般都采用多项目定标液,一瓶定标液可以定标多达20几个项目,这20几个项目用这一瓶定标液就可以搞定了。
当然多点定标的免疫比浊项目不行,是单独的定标液。
黄色架子的第一个位置要放纯水,告诉电脑这是零浓度点,也就是第一定标液。
第二个位置才放多项目定标液,告诉电脑不同的项目的值是多少。
其后才是放置质控品的绿色架子和常规样本的白色架子。
很多时候由于定标液太多,特别是开了不少多点定标的项目,一个黄色架子排不完。
那就多排几个黄架子,如果没有,就一个黄架子,那就多走几遍。
第一个黄架子之后跟着第一个黄架子有关的质控绿架子,然后测试,就不要放白架子了。
这一遍走完,把没有做的定标液换上,再跟与之有关的质控绿架子,再来一遍,直到定标和质控完全走完再走常规样本。
很多定标问题都出现黄架子的第一个位置是定标液,根本没有水点,理由是前面的兰架子做了试剂空白了啊?
!
这是典型的误区。
还有的不放水点的原因是黄架子位置不够了,没法做。
这是不是理由的理由,搬家一次搬不完不会分两次啊,一口吃成胖子不成?
总结:
线性定标最好选择两点,其中可以包括水点(零浓度点),坚决摈弃K因数法。
3.2多点线性定标:
很少用,我也不知道什么项目会用到。
两点决定一线了,还要那么多点干吗呢?
只知道同工酶法是多点线性,但如何应用并不清楚。
不过存在必有道理,我孤陋寡闻罢了。
3.3非线性定标。
针对线性定标的直线来说,非线性定标肯定是曲线,曲线的趋势不同,决定了不同的结果走向。
非线性定标最少需要2个点,但一般最少选择4个点,否则曲线难看不说,结果也做不好。
非线性定标大致分为对数(logit)曲线,折线(polygonal),指数(exponet)曲线和样条(spline)曲线。
下图是对数曲线
下图是样条曲线:
下图是折线
下图是指数曲线
折线和指数曲线很少使用,我还没在应用中见到过用这两种曲线的定标项目。
奥林巴斯和拜耳把所有的非线性定标都认作是多点定标,至于曲线趋势根本不管你是对数还是样条,只要你给我定标点,我就给你画出来,然后再定标曲线上查找对应的值。
但奥林巴斯的多点定标又有ACAL和MCAL之分。
ACALn(2-7)AB是自动进行多点定标,最少要有2个点。
但这个软件人机对话方面有些问题,按理说按照日立的设计,多点定标分那么种,你都给罗列上不就行了吗?
不,奥林巴斯分了两步进行输入,首先输入定标类型,也就是多点定标还是单点定标,线性还是非线性,自动还是人工。
然后在后面紧跟着一个公式选择,让你选择计算公式是直线方程还是对数、折线、样条等等方法。
这下把操作人员又搞糊涂了。
上图是多点定标的曲线,看起来也是对数曲线,下图是定标设置的参数屏幕
前面的红圈是选择定标方式的,多点定标的话最少要选择2AB以上,图上是5AB,也就是5个定标点。
后面的红框是选择计算公式的,图上选择的是折线(polygonal),但是很明显,CRP用折线很不合适。
前面的定标点选择也就是定标方法选择很少出现问题,有几个定标液大家都知道,往往是后者公式选择错误,造成定标质控还可,结果一塌糊涂,特别是高低值的标本。
我还真记不住公式里面都有什么,记得有直线方程,折线,样条,对数和指数。
再看上图,CRP的5个定标点没有水点,也就是说没有零浓度点,那么系统会默认从原点开始。
这就是很多人做出来的非线性曲线在最低浓度处会出现一个怪异的扭曲的原因。
而奥林巴斯的MCALn(1-7)MB是人工多点定标,与自动多点定标的区别是人工多点定标认为是折线,每一个线段都有一个斜率,做不出曲线来。
所以这一点也要注意。
对数曲线大家都很数学,中学学习对数的时候没少折腾,俗称抛物线。
日立和东芝把对数曲线分的很细,有LOGIT-LOG3P/4P/5P等,分别代表不同的对数趋势,所用的定标点都一样,2-6个。
Log3P的趋势图
Log4P的趋势图
Log5P的趋势图
从这里可以看出不同的Log趋势,对相应高低浓度的吸光度对应有着不同的对应,所以在方法没有错误的情况下,结果高值不高,低值不低,甚至相反的时候,可以在这些机型中找到对应的曲线趋势进行重新定标。
有人说只要是多点定标就采用样条Spline,这仅仅是大多数而已,也有特例的。
对数与样条曲线的差异还是很明显的。
一般来说,除非厂家明确给出定标方法,如果没有给出,多点定标先选择样条曲线进行定标,定标后观察曲线,人工判读是折线好,还是对数好,还是就是样条。
对数的话还要看是哪一种对数。
要有一个人工判读修改的过程,不能什么都交给机器,机器毕竟是死的,你告诉它什么,它就认为是什么。
多点定标中,理论上讲应该倍比稀释,也就是说厂家发来一瓶定标液,要倍比稀释5份,加上一份水点,按照浓度从低到高的顺序加入测试进行定标。
这也是最经济的办法,毕竟一瓶定标液的价钱在哪儿摆着。
例如一瓶标有浓度为40的定标液,倍比稀释加上水点后的浓度依次为0、2.5、5、10、20、40(0、1/16、1/8、1/4、1/2、1),这样做的定标方法可以直接选择样条曲线。
但也有很多厂家给出了五种定值的校准品,而且并非倍比的。
相邻定标值不是两倍关系,甚至多达几十倍的关系,这样做的目的无非是要拉宽测试结果范围,能做出很高的值来。
但是这样一来,超高值的可靠性就很令人怀疑,而且肯定做不准的。
我听到过很多人对此的解释是反正已经超高了,准不准的意义不大。
唉,这也是国情所在,毫无办法。
老老实实做事的,被冠以结果范围太窄,稍微一高就做不出来,要人工稀释才行。
而这些用这种方法做出来的,根本无人管结果准不准,只要是能减轻他的工作强度就行。
如果采用了多种定值校准品,而且不是倍比关系的,就不能采用样条方式,在对数曲线里找一种合适的吧,或者看看指数函数是否适用。
一味拉高定标值也源自机器设置,超过定标最高值,就不出结果,提示超过定标值,这样引起操作人员的不满。
唉,稀释啊,都有LIS,稀释后传到LIS上,在LIS上修改一下结果。
但这会加重操作人员的负担,哪怕是计算器算一下,手工稀释一下,再做几遍的时间。
一般遇到多种定值校准品校准后结果出现问题的,我都要求用最高值倍比稀释重新定标,方法还是采用样条曲线。
而且多次试验证明,用最高值倍比稀释后,再重测其它四种定标液,得出的数值有的相差甚远,厂家当初给的值就不准,怎么能保证结果的准确呢?
总结:
多点定标别忘了水点,倍比稀释才能保证结果准确,在无法判定方法的情况下,先采用样条曲线,然后人工判读修改。
3.4在定标中,有很多检查需要注意。
如空白吸光度,每一点的吸光度范围检查,线性检查,灵敏度检查等等,如果设置了这些检查,那么超过这些检查值就会报错,定标不会通过。
同样,通过了这些检查的定标肯定是值得信赖的。
如果不想进行检查,要想定标顺利通过,那最好取消或者将这些值放到最大。
这样做定标虽然会通过,但准确性大打折扣,自己心里都没有底儿。
定标后的校正问题:
定标后特别是多点定标后,对曲线不满意,或者样本结果不满意,可以采取重新定标(也叫全点校正)或者单点多点校正方式修正定标曲线。
通俗的讲就是取其中一个点或一两个点进行定标,从而修正整条曲线的方法。
采取这种情况,一般是多点定标液的一种或几种用完了,或者只有其中的一两种的情况下。
除非必要,否则真的没理由采取这种方式。
下面的图示是奥林巴斯的单点校正示意图
下图是东芝的单点和两点校正示意图
说白了,就是根据原来的曲线趋势,任意改变其中的一点或两点,自动进行其余点的浓度计算,从而修正整个曲线。
我有个朋友是做应用的,他最喜欢干的事情是做单点或两点校正,认为这样省事省力。
但经常出现结果还不如从前呢。
于是乎我下班回家做饭,这位老兄下班呢到我家吃饭,然后就写写画画,分析今天的单点校正为什么不行,昨天的两点校正为什么那么怪异。
我的反应一向很慢,在数据不充分,没有查询的情况我很难得出一个令我自己信服的结论。
我信奉一个原则,那就是省事儿就是费事。
所以我从不做单点或两点校正,而是全点重新定标。
每次看到老兄给我的曲线及后面的校正图后,都能找到相应的校正点,而不是他选择的。
也就是说出问题的点不是他修正的那个点。
这个判断很难描述,特别是在没有案例曲线的情况下。
这位老兄也云山雾罩,死活不理解我的判断。
后来,大概半年后,这位朋友彻底抛弃了单点或多点校正的方法,改为老老实实的全点定标,真正的省事省力了。
本主题由郑振寰于2014-6-1915:
44加入精华
窗体顶端
post_newreply
窗体底端
分享到:
QQ好友和群 QQ空间 腾讯微博 腾讯朋友
收藏12 分享 微信
看贴要回是本分,有问必答是人才,解决问题回贴
 升级会员
升级会员 