引物设计流程之基因编码区CDS扩增引物设计Word文档格式.docx
《引物设计流程之基因编码区CDS扩增引物设计Word文档格式.docx》由会员分享,可在线阅读,更多相关《引物设计流程之基因编码区CDS扩增引物设计Word文档格式.docx(20页珍藏版)》请在冰点文库上搜索。
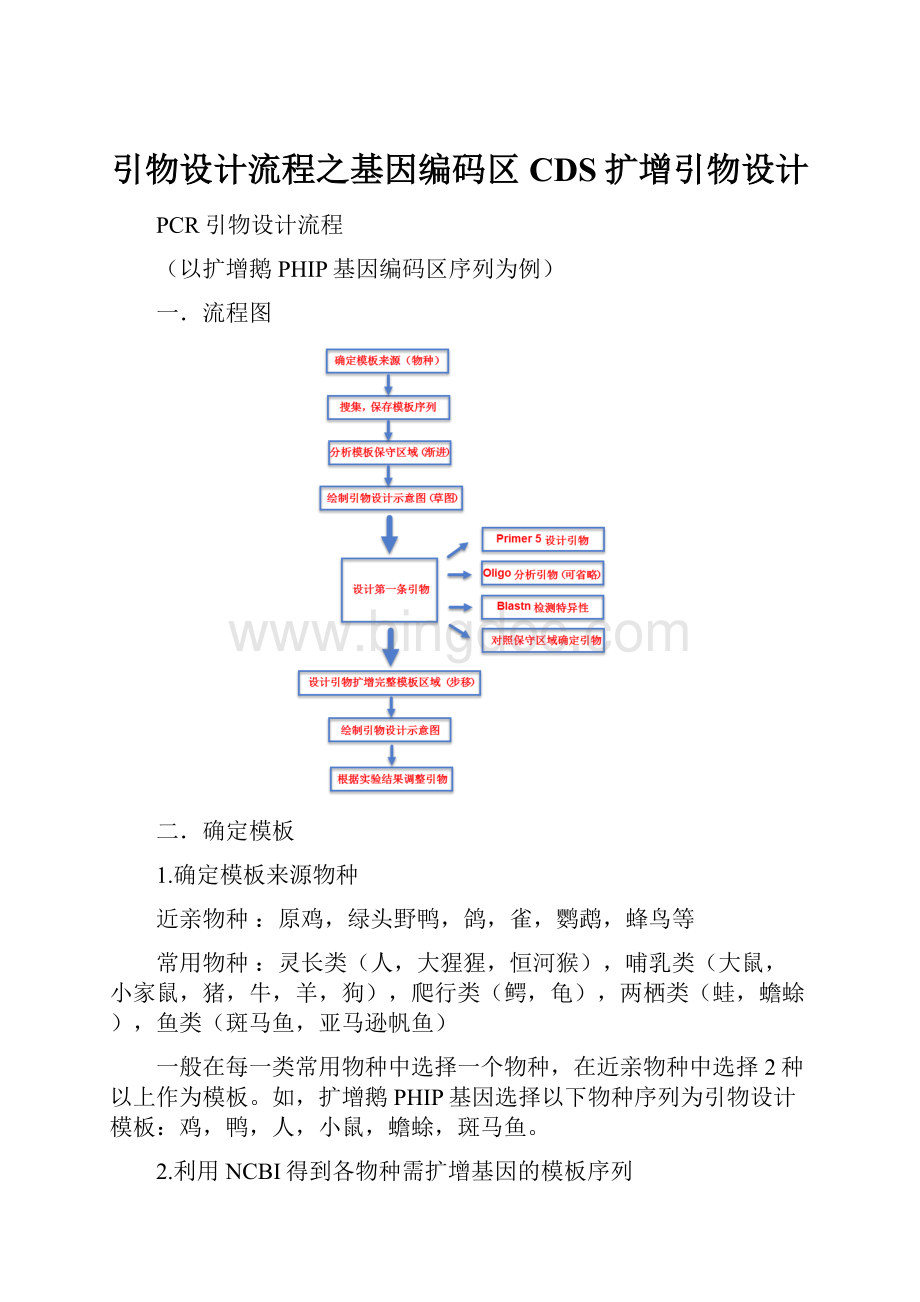
C.需注意:
对于同一基因的mRNA可能具有不同长度的剪切异构体,选择模板时不同物种应尽量选择同一异构体(一般选择最长的异构体)。
D.如需得到该基因所在基因组的序列信息(如扩增启动子区域时),在基因报告页面上部Genomicregions,transcripts,andproducts条目下,点击Gotonucleotide选项下FASTA按钮可进入基因组(组装)序列页面。
E.在基因组(组装)序列页面中,默认仅显示跳转前基因的序列,在Changeregionshow条目中修改设置为Wholesequence得到基因组序列,在Send选项下保存即可。
3.整理下载的模板序列
三.寻找保守区域
保守区域的意义:
基因的保守区域是指不同来源的同一个基因在某些区域没有差别或者差别很小。
在扩增基因序列时,选择在保守区域设计引物能够更有效的扩增未知的基因。
因此,在引物设计前需先找出目的序列中的保守区域。
在引物设计时,则首先应在保守区域内设计引物。
1.制作mega标准序列
A.用ClustalX软件打开整理好的TXT文件(菜单File→LoadSequences),然后在菜单Alignment选项下选择DoComplementAlignment,此时将保存两种格式文件:
.dnd和.aln(序列较长时耗时较长,需数分钟)。
B.将以上.aln文件用DAMBE软件打开(File→Openstandardsequencefile),注意选择恰当的序列类型。
打开后的文件可直接转存为.meg或.fas格式文件(File→SaveorConvertSequenceFormat)。
注1:
此时在SequenceInfo对话框应选择Binary选项,否则在转换格式时T碱基会被替换为U。
2.Mega分析同源性
A.用Mega软件打开以上保存的.meg或.fas格式文件,新建一个序列对齐分析窗口(Align→Edit/BuildAlignment→Retrievesequencesfromafile)。
B.在新窗口中以默认设置将序列集进行序列对齐分析(Alignment→Alignbymuscle(Codons)),结果保存为.fas文件(Data→ExportAlignment)。
C.打开网址http:
//mobyle.pasteur.fr/cgi-bin/portal.py#forms:
:
boxshade进入在线软件BOXShade,在选择文件按钮下载入以上已对齐的模板.fas文件。
点击advancedoptions按钮展开选项,将默认设置进行如下图修改。
其余选项保持默认即可,也可根据实际需要进行调整。
设置完后点击Run提交任务。
注2:
shouldsequencenamebeprinted项的默认设置为YES,此时将把序列名同时打印。
需再输出的富文本文件中将字体全部改为中文字体,才能保持序列的对齐。
如需英文字体,此项可以改为no,序列名可以通过其他方式添加。
在软件输出结果的报告页面中保存.rtf格式的富文本文件,该文件可用MicrosoftWord编辑。
3.保守区域的分析
如下图,consenus行表示序列间的一致性,*号表示在序列间完全一致的碱基,.号表示在序列间高度相似的碱基,空格表示在序列间;
consenus行之上每一行分别代表一个物种的PHIP基因序列,蓝色背景的碱基为在物种间保守的碱基。
首先观察consenus行,*号比例在50%以上且空格很少的区域可视作保守区域,具体分析中应灵活处理。
以PHIP基因为例,保守区域可划分为:
190-1240bp,1260-1980bp,2000-2290bp,2540-4920bp,5340-5740bp。
注3:
出现连续的长的不保守区域(大于数百bp),因为引物设计产物的最佳长度上限在1000左右。
此时可以只考虑近亲物种序列的保守性,降低保守区域的分析标准。
4.绘制引物设计示意图
注4:
对于PHIP基因,我们顺利的得到了保守性区域分析结果。
但是,对于某些进化较快的基因,保守性区域可能不足够用于设计引物。
此时,可以逐渐减少用于分析的远缘物种,采用渐进性的分析,直到得到能够设计引物的保守性区域。
注5:
为什么是mega?
保守性区域的分析用其他软件(如DNAMAN)也可进行。
采用mega的原因在于其分析方法的可靠性更高,同时该软件在进化分析等中也非常常用,所以将mega可读序列的制作一并说明。
四.用Primer5软件设计引物
1.新建文件,导入模板
确定一条序列为设计引物的模板序列。
本例中根据进化关系,选择鸭的PHIP基因序列为模板。
在Primer5软件中新建一个窗口(File→New→DNASequnce),将模板序列粘贴(ctrl+v)在窗口内(一般选择asis表示粘贴原序列,也可根据需要粘贴反向序列等)。
2.设置参数,搜索引物
在新序列窗口中点击
按钮进入引物设计窗口,如下图。
在引物设计窗口中点击
按钮进入引物搜索窗口,如下图。
引物类型为PCRPrimers,搜索类型一般选择成对引物,在搜索范围内限制搜索上下游引物的序列区域(参考前面的保守区域进行设置),产物长度,引物长度设置详见后续介绍。
搜索模式分为自动的Automatic和手动的Manual,在自动模式下引物搜索由严格标准往宽松标准执行,直至引物条数/对数达到设定值,其搜索参数设置如下图,搜索参数为在搜索过程中排除不合格引物的筛选条目;
在手动模式下,可设置搜索的严格程度,并可修改搜索参数条目下的限定数值。
在引物搜索窗口设置好后点击
按钮开始搜索,搜索结果如下图。
在搜索结果中可分别查看上下游引物或成对引物的情况。
Rating值代表系统对引物的(产物)打分,分值越高说明引物越优秀,但并不是绝对的评价标准。
引物具体信息在点击引物所在行后在引物设计窗口显示,其中:
按钮为选择显示上游或下游引物信息;
SeqNo表示引物第一个碱基在序列中的位置;
Length表示引物/产物长度;
Tm表示引物/产物的熔解温度;
GC%表示引物/产物的GC含量;
△G表示引物结合模板过程的自由能;
Activity表示引物与结合的效率;
Degeneracy表示引物的多义性;
TaOpt表示Primer5软件建议的成对引物扩增时的最佳退火温度。
Hairpin表示引物可能形成的二级结构;
Dimer表示引物自身可能形成的二聚体;
FalsePriming引物与模板的错频;
CrossDime引物间可能形成的二聚体表示。
以上项目即为分析引物时的参考条目,引物设计的一些原则整理见后续。
参照引物设计的原则即可根据Primer5中上述参数对引物对进行选择。
另,对于一些引物,可能出现大多数指标都较为优秀,但个别指标严重影响扩增反应的情况。
这种引物可使用
按钮,手动的对其进行修改以提高其性能。
3.引物设计原则
1)引物长度:
一般为15-30bp,常用的是18-27bp,但不能大于38,因为过长会导致其延伸温度大于75℃,即Taq酶的最适温度。
总的说来,每增加一个核苷酸引物特异性提高4倍,这样,大多数应用的最短引物长度为18个核苷酸。
引物长度的上限并不很重要,主要与反应效率有关。
由于熵的原因,引物越长,它退火结合到靶DNA上形成供DNA聚合酶结合的稳定双链模板的速率越小。
2)产物长度:
扩增片段长度取决于酶的活性和保真性能。
对于普通Taq聚合酶,PCR产物一般不超过2000bp,而在100-1000bp范围效果较佳,超过1000bp的产物就可能出现产物量降低甚至无法扩增的情况。
对于其他酶,应根据相关说明使用。
3)引物Tm值:
引物的Tm值,指的是50%的引物分子和其互补序列表现为双链时的温度,PCR时的退火温度一般都要比Tm值低5℃左右以确保有效退火。
引物的Tm值一般控制在55-65度,一般需保证上下游引物的Tm值差不超过4-6度。
如果引物中的G+C含量相对偏低,则可以使引物长度稍长,而保证一定的退火温度。
许多软件可以对Tm进行计算,其计算原理各有不同,因此有时计算出的数值可能会有少量差距。
4)GC%:
有效引物中(G+C)的比例为40-60%,GC含量太低导致引物Tm值较低,使用较低的退火温度不利于提高PCR的特异性,GC含量太高也易于引发非特异扩增。
上下游引物的GC含量不能相差太大。
GC%对扩增的影响主要通过Tm值来体现,当Tm值符合要求时,对于GC%不必做严格要求。
另,引物序列中同一碱基连续出现不应超过5个。
5)引物3’端:
引物3’端是延伸开始的地方,最好不存在错配。
同时3’端不应超过3个连续的G或C,因这样会使引物在G+C富集序列区错误引发。
同时,3’端有形成二级结构/二聚体的可能对于PCR扩增的影响将大于5’端。
在扩增编码区域时,引物3′端最好不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增特异性与效率。
6)△G值:
引物5′端和中间△G值应该相对较高,而3′端△G值较低。
△G值是指DNA双链形成所需的自由能,它反映了双链结构内部碱基对的相对稳定性,△G值越大,则双链越稳定。
应当选用5′端和中间△G值相对较高,而3′端△G值较低的引物,即3’端尽可能选用A或T,少用G或C。
引物3′端的△G值过高,容易在错配位点形成双链结构并引发DNA聚合反应(寡核苷酸3′末端最后5个核苷酸的稳定性小于-9kcal/mol,通常就是专一性的探针或引物)。
7)引物的二级结构/二聚体:
引物自身不应存在互补序列,否则引物自身会折叠成发夹状结构,这种二级结构会因空间位阻而影响引物与模板的复性结合。
另外,尽可能避免两个引物分子之间3’端有有较多碱基互补,否则两个引物分子可能通过互补碱基而结合形成引物二聚体。
一般情况下,引物形成的发夹结构和二聚体中不应多于4个连续碱基的同源性或互补性(实际常用自由能△G判断,△G为负值时,其绝对值越大不利影响越大,一般以小于绝对值4.5为标准(应远小于引物与模板结合的△G),为正值时则几乎无影响)(Oligo分析时的一般标准则为引物效率小于100或小于目的产物效率的三分之一)。
个人观点认为,引物的二级(发夹)结构对于目的片段扩增的不利影响要大于引物二聚体(因为引物位置的二级结构在模板序列中是同样存在的。
模板序列形成二级结构,在PCR过程中如果无法完全解链则将直接导致引物与模板结合的失败)。
8)引物5’端:
可以有与模板DNA不配对碱基。
其应用有:
5’端加上限制性核酸内切酶位点序列(酶切位点5’端加上适当数量的保护碱基),5’端的某一位点修改某个碱基,人为地在产物中引入该位点的点突变以作研究,5’端标记放射性元素或非放射性物质(如生物素、地高辛等)等。
因此,在无法避免的情况下,引物5’端的出现的错配在一定程度上可以接受(也用自由能△G判断,标准与“引物的二级结构/二聚体”条目相同)。
4.保存引物
通过File→Print可以保存引物的报告文件.pdf(需安装虚拟打印机)。
注6:
以上是Primer5使用的基本流程。
更详细的说明参照本文相关文件夹内“PrimerPremier5.0中文使用说明书”文件。
注7:
部分资料中对于引物形成二级结构的预测提到了用单独的软件进行检测(如RNAStructure)。
该软件可在其官网网址免费注册后下载http:
//rna.urmc.rochester.edu/register.html,也可在线使用http:
//rna.urmc.rochester.edu/RNAstructureWeb/。
其使用说明可参照官方网站,也可参照本文相关文件夹内“RNAStructure3.2中文使用说明书”文件。
注8:
在引物设计软件中,Oligo软件也是一款很常用的软件。
其软件的使用技巧,详见本文相关文件夹内“使用Oligo6和PrimerPremier5.0等软件设计PCR引物”文件。
一般情况下,单独使用一个软件已足够;
本文建议使用Primer搜索和筛选引物后,备选引物在Oligo中逐一分析。
注9:
引物设计是一项复杂的工作,限于篇幅和时间等原因,本部分的介绍还很粗略。
在本文相关文件夹内“引物设计相关论文”文件夹中有一些关于引物设计的论文(大多选取来源于CSCD等数据库的优秀期刊),是对本文内容的补充。
同时,大家也可以自行下载和搜集更多的论文和资料,希望有精力的同学可以继续完善和提升本文。
五.用NCBIBlastn程序比对引物
目的:
检测设计的引物是否在其他非目的基因(模板)上存在非特异性的结合。
1.Blastn的使用
由网址http:
//blast.ncbi.nlm.nih.gov/Blast.cgi?
PROGRAM=blastn&
PAGE_TYPE=BlastSearch&
LINK_LOC=blasthome进入Blastn在线程序。
在EnterQuerySequence条目下输入或粘贴备选的引物序列。
在ChooseSearchSet条目下Database中选择搜索的数据库,一般选择Nucleotidecollection数据库。
其余设置为默认值。
注10:
选择Nucleotidecollection数据库原因在于其包括的序列完整,既包括预测序列也包括实验获得序列。
同时,该数据库并未区分dna序列和mRNA序列。
因此,在分析比对结果时应根据引物设计目的进行分析(扩增dna序列时,引物与mRNA的非特异性结合对扩增并无影响;
反之亦然)。
ProgramSelection条目下可选择比对的严格程度,建议选择最低的严格程度(Somewhatsimilarsequences),因为在结果页面将按序列的相似性由高到低排列比对结果,如果选择高的比对标准可能导致漏掉对PCR扩增有较大影响的比对结果。
其余设置一般保持默认,在页面下部点击
按钮即开始比对。
2.结果分析
Blastn的结果输入页面主要包括三个部分。
第一部分是图形式的汇总,用于初步了解给定序列与数据库序列的匹配情况。
第二部分是列表式的描述。
覆盖率即是给定序列与数据库序列形成的匹配占给定序列长度的比例。
第三部分是逐个显示详细的匹配情况。
匹配率即匹配序列中匹配碱基的比例。
匹配数量是指两段匹配序列能可能的匹配方式,给定序列可能与数据库序列的不同区段发生匹配,因此该数量可能大于1。
Blastn进行序列比对时将自动考虑序列的方向,并同时考虑序列和互补序列,因此查看结果时可能需要查看“Strand”项,并查看“序列匹配情况”中的碱基数标号以确定两端序列匹配的方式(匹配,反向匹配,互补匹配,反向互补匹配)。
3.将上下游引物均进行以上分析,保存错配模板信息,将上下游引物与目的物种或近亲物种中同一非目的模板存在匹配的引物对剔除,处理后如剩余引物对较多,在只存在单向引物错配的序列中灵活处理和剔除(该尺度可比设计引物时考虑“错配”时的标准适当放宽)。
注11:
以上是Blastn的简单使用说明,详细介绍参考ftp:
//ftp.ncbi.nlm.nih.gov/pub/factsheets/HowTo_NewBLAST.pdf。
六.备选引物的保守性检查
已设计好的备选引物应该再次对照基因的保守区域分析结果,根据其具体的保守性情况,在不影响扩增性能的情况下,选择保守性较好的引物。
七.完整编码区的引物设计
完成一段模板的引物设计后即可以进行下一段引物的设计,直至完成全部序列的引物设计。
在设计下一段引物时应注意与上一段引物扩增区域保留一段重叠序列(至少需要数十bp,条件允许时最好在100-200bp。
尤其在每段引物扩增模板序列长度较长时,因为测序时的末端序列常易出现可靠性降低的情况),以便于完整序列的拼接。
以PHIP基因为例,其编码区5端前200bp序列保守性较差。
对于编码区两端序列保守性较差的情况,可根据该物种的基因组序列,参考其他物种的基因组序列或具有完整mRNA序列的物种,寻找基因两端UTR(非翻译区)中保守区域设计跨域起始/终止密码子的引物。
如果由于模板质量原因,或是无法设计设计跨域起始/终止密码子的引物时,也可先设计引物扩增基因的dna序列,确认起始/终止密码子附件的序列后,再设计引物扩增mRNA序列。
(对于序列两端保守的基因则可以直接将第一个和最后一个引物设计在起始/终止密码子上)。
建议在完成完整编码区的引物设计后再绘制一个具体的引物设计示意图(其他区域的扩增也是一样)。
注12:
在引物设计出现困难时,也可考虑进行简并引物的设计。
简并引物设计的说明详见本文相关文件夹内“简并引物设计”文件内,其相关论文见“简并引物相关论文”文件夹内。
简并引物设计的具体方法暂未见详细的介绍,有精力和兴趣的同学可以进行搜集和整理。
 升级会员
升级会员 